Research Focus |

| |
|
使用胰腺肿瘤类器官库(PTOL)检测Wnt信号作为胰腺导管腺癌(PDAC)的治疗靶点 2024-04-17 20:13:31 浏览次数:2393 | |
| 使用胰腺肿瘤类器官库(PTOL)检测Wnt信号作为胰腺导管腺癌(PDAC)的治疗靶点 来源:仪方生物 www.yeslab.com 胰腺导管腺癌(PDAC)表现为晚期,对大多数治疗方式都是难治性的。Wnt信号激活在增殖和化疗耐药性中起着关键作用。最小培养基条件、生长因子依赖性和Wnt依赖性是通过对来自胰腺肿瘤类器官库(PTOL)的七个患者衍生类器官(PDO)的Wnt抑制来确定的。使用患者来源的异种移植物在体内评估在体外显示反应的类器官。鉴定了每个类器官的Wnt(in)依赖性基因特征。与单独化疗或ETC-159相比,当用ETC-159与紫杉醇或吉西他滨联合治疗时,Panc269显示出类器官生长减少的趋势。Panc320在ETC-159和紫杉醇的组合中显示出更显著的抗增殖作用,但在吉西他滨中没有。将Panc269和Panc320植入裸鼠体内,并用ETC-159、紫杉醇和吉西他滨作为单一药物和联合治疗。ETC-159和紫杉醇的组合显示出比单独的ETC-159更大的抗肿瘤效果。在Panc320异种移植物中观察到了较小程度的联合治疗效果。Panc269和320的Wnt(in)依赖性基因特征与所显示的表型一致。通过RT-PCR评估的几个关键Wnt基因的基因表达在体内处理后显示出显著的倍数变化。每种胰腺类器官都表现出不同的小生境因子依赖性,为靶向治疗提供了途径,并通过Wnt抑制剂联合治疗和体外标准化疗后的生长分析得到支持。迄今为止,在我们用Wnt抑制剂加紫杉醇或吉西他滨治疗的患者衍生异种移植物模型中,这种联合治疗模式在胰腺癌症PDO中的临床应用得到了支持。基因表达分析表明,有一些关键的Wnt基因有助于胰腺肿瘤的Wnt(in)依赖性表型,在基因型水平上为Wnt(n)依赖性和治疗易感性或耐药性提供了合理的机制解释。 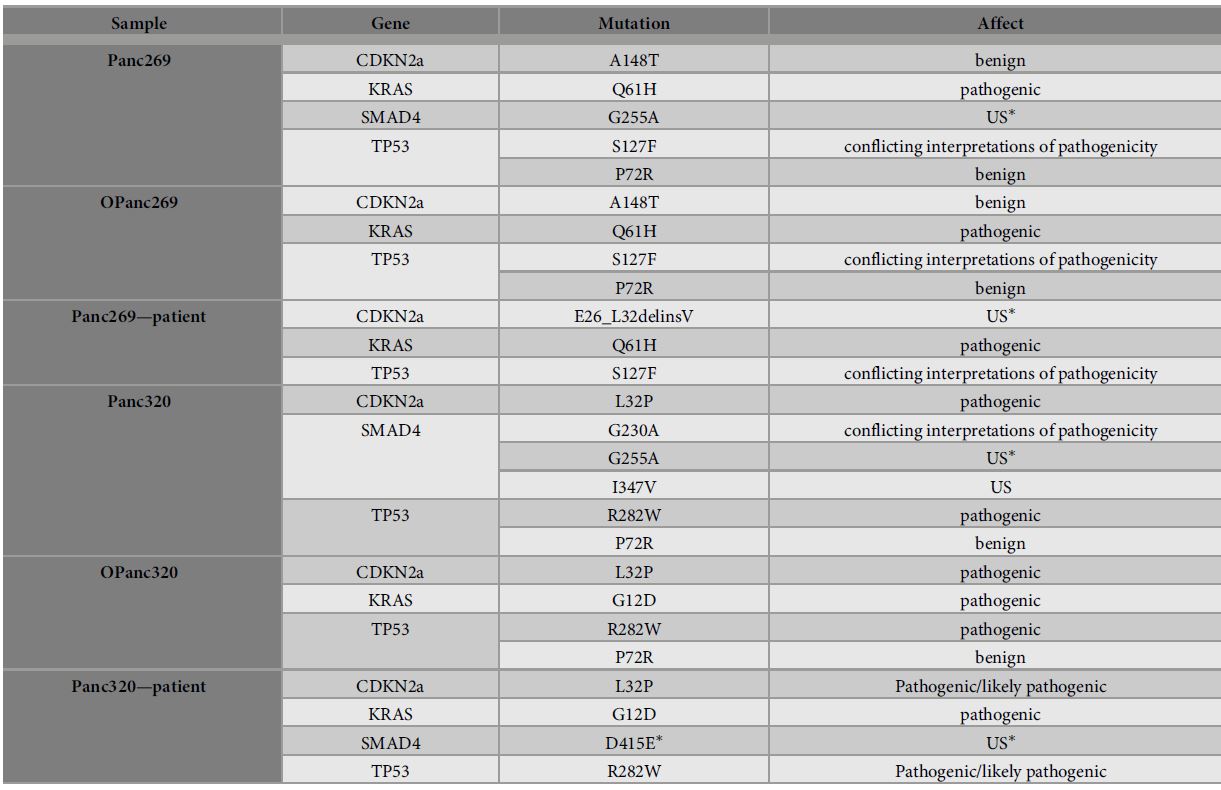 胰腺导管腺癌(PDAC)目前是癌症死亡的第四大常见原因,其5年生存率低于8%,这是由于其在晚期表现的趋势,而对大多数治疗方式来说是难治的[1]。关键癌基因和抑癌基因,如KRAS、TP53、SMAD4和CDKN2a中的许多突变和体细胞拷贝数改变(SCNA)导致PDAC[2,3]的发展。除了这些基因改变外,癌症基因组图谱研究网络还揭示了PDAC和染色体重排模式更复杂的分子景观[4]。这突出了在评估肿瘤的基因型和生物反应之间的关系时出现的困难。胰腺癌中常见的低肿瘤细胞数和致密基质进一步混淆了这些关系,并成为准确有效的临床前研究的主要障碍[5,6]。 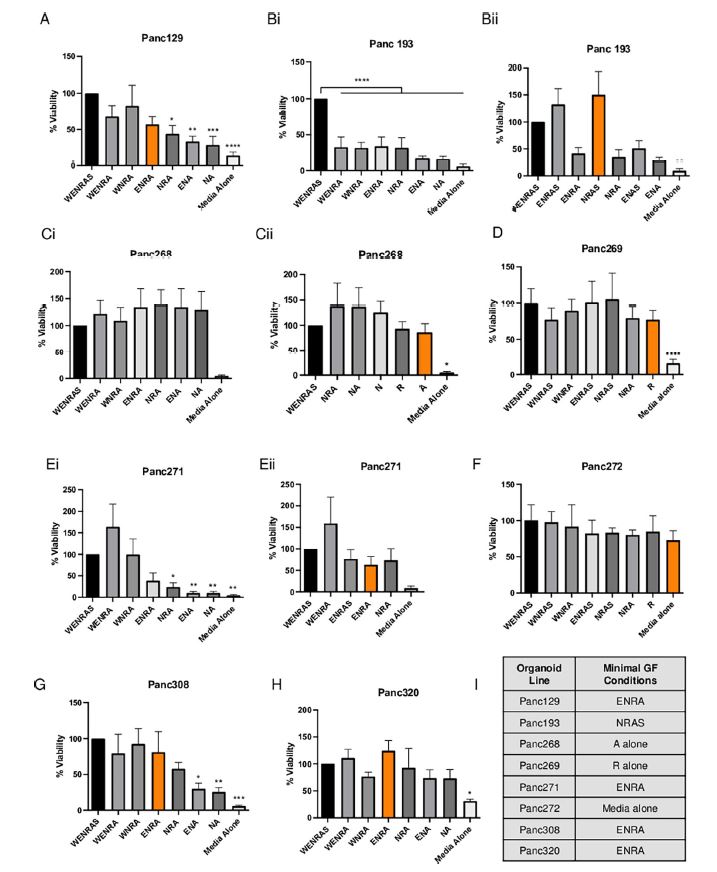 尽管Wnt激活突变在PDAC中很罕见,但Sato小组最近的观察结果和我们自己使用PTOL识别Wnt依赖性PDAC亚型的初步数据表明,Wnt依赖可以在克服化疗耐药性的治疗策略中提供信息。因此,本研究的目的是确定Wnt依赖性是否能预测Wnt抑制剂的反应并逆转化疗耐药性。 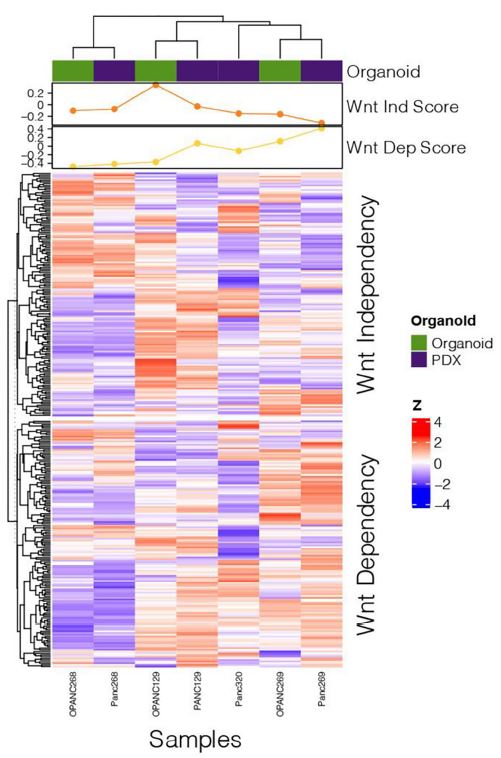 对我们原始患者肿瘤的突变分析揭示了几个先前发现的显著的关键驱动突变。鉴于这些临床突变数据,利用全外显子组测序来评估本研究中使用的PDAC类器官的遗传突变谱。对癌症发展的主要贡献者尤其受到重视,即TP53、KRAS、CDKN2a和SMAD4[2,3]。我们检查了由Panc268、Panc269、Panc320和各自的类器官产生的序列中这些基因的突变。表1显示了根据ClinVar[40]鉴定的致病突变。  PTOL肿瘤的临床突变图谱显示,癌基因(如KRAS)和肿瘤抑制基因(如TP53)突变的频率与一般胰腺癌症人群相同(表1)。例如,Panc269表达常见的KRAS和TP53基因突变,而Panc272没有可检测的基因突变。这与下面确定的生态位因子依赖性无关。 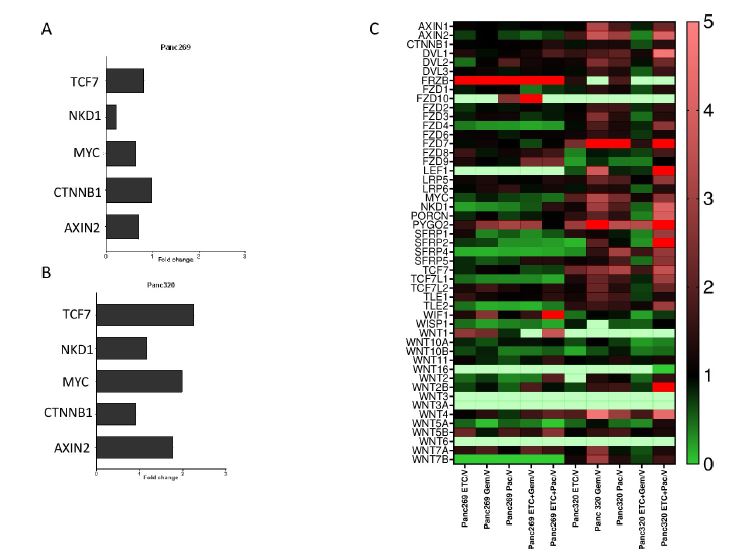 正如这项研究所强调的,基因型和生物表型很难关联起来,从而实现准确的相关性[6]。虽然我们已经调整了类器官的利用,以帮助弥合基因型和表型之间的差距[29],但当将体外数据应用于更临床的环境时,这一差距仍然是一个挑战。我们能够根据Wnt信号传导以及对抑制和化疗的反应有效地对PDAC类器官的表型进行分类。在考虑我们数据背后的机制时,与基因型的关系问题仍然是一个关键问题。除了获得的PCR数据外,对Wnt依赖性和Wnt非依赖性基因的热图确定了大多数Wnt非独立类器官的Wnt独立基因特征,以及大多数Wnt依赖类器官的W依赖性基因特征,这与佐藤小组之前的工作一致[1]。一致的基因特征可用于更有效地对胰腺肿瘤中的Wnt(in)依赖性进行分类。PDAC PTOL基因特征的这种分类可作为预后指标,用于考虑将Wnt抑制治疗与标准化疗相结合,用于那些依赖Wnt信号传导生长的肿瘤。它还可以作为一种工具,更好地预测患者衍生的胰腺类器官的预期表型。此外,我们需要验证类器官与其互补PDX之间的差异。这可能包括将各种生长因子引入小鼠,以补偿肿瘤微环境与类器官生长介质的差异。 这项研究在很大程度上受到我们的PDAC类器官库的大小和这些类器官的效用的限制,这些类器官由不同的生长速率决定。为了更普遍地接受结果,这些实验需要在开发过程中对更多的类器官进行。 Examination of Wnt signaling as a therapeutic target for pancreatic ductal adenocarcinoma (PDAC) using a pancreatic tumor organoid library (PTOL) Pancreatic ductal adenocarcinoma (PDAC) presents at advanced stages and is refractory to most treatment modalities. Wnt signaling activation plays a critical role in proliferation and chemotherapeutic resistance. Minimal media conditions, growth factor dependency, and Wnt dependency were determined via Wnt inhibition for seven patient derived organoids (PDOs) derived from pancreatic tumor organoid libraries (PTOL). Organoids demonstrating response in vitro were assessed in vivo using patient-derived xenografts. Wnt (in)dependent gene signatures were identified for each organoid. Panc269 demonstrated a trend of reduced organoid growth when treated with ETC-159 in combination with paclitaxel or gemcitabine as compared with chemotherapy or ETC-159 alone. Panc320 demonstrated a more pronounced anti-proliferative effect in the combination of ETC-159 and paclitaxel but not with gemcitabine. Panc269 and Panc320 were implanted into nude mice and treated with ETC-159, paclitaxel, and gemcitabine as single agents and in combination. The combination of ETC-159 and paclitaxel demonstrated an anti-tumor effect greater than ETC-159 alone. Extent of combinatory treatment effect were observed to a lesser extent in the Panc320 xenograft. Wnt (in)dependent gene signatures of Panc269 and 320 were consistent with the phenotypes displayed. Gene expression of several key Wnt genes assessed via RT-PCR demonstrated notable fold change following treatment in vivo. Each pancreatic organoid demonstrated varied niche factor dependencies, providing an avenue for targeted therapy, supported through growth analysis following combinatory treatment of Wnt inhibitor and standard chemotherapy in vitro. The clinical utilization of this combinatory treatment modality in pancreatic cancer PDOs has thus far been supported in our patient-derived xenograft models treated with Wnt inhibitor plus paclitaxel or gemcitabine. Gene expression analysis suggests there are key Wnt genes that contribute to the Wnt (in)dependent phenotypes of pancreatic tumors, providing plausible mechanistic explanation for Wnt (in)dependency and susceptibility or resistance to treatment on the genotypic level. Pancreatic ductal adenocarcinoma (PDAC) is currently the fourth most common cause of cancer death with a 5-year survival rate of less than 8% due to the tendency of presentation at an advanced stage while being refractory to most treatment modalities [1]. Many mutations and somatic copy number alterations (SCNAs) in key oncogenes and tumor suppressor genes, such as KRAS, TP53, SMAD4, and CDKN2a, lead to the development of PDACs [2, 3]. In addition to these genetic alterations, the Cancer Genome Atlas Research Network has revealed an even more complex molecular landscape of PDACs and chromosomal rearrangement patterns [4]. This highlights the difficulties that arise when assessing the relationship between the genotype and biological responses of tumors The low tumor cellularity and dense stroma commonly found in pancreatic cancers further convolute these relationships and serve as major barriers to accurate and effective preclinical studies [5, 6]. Although Wnt activating mutations are rare in PDACs, recent observations by Satos group and our own preliminary data using PTOLs to identify Wnt dependent PDAC subtypes suggest that Wnt dependency can be informative in therapeutic treatment strategies to overcome chemotherapy resistance. Thus, the goal of this study was to determine whether Wnt dependency will predict Wnt inhibitor response and lead to reversing chemotherapy resistance. Mutational analysis from our original patient tumors revealed several notable key driver mutations as previously identified. Given this clinical mutation data, whole exome sequencing was utilized to evaluate the genetic mutational profiles of the PDAC organoids used in this study. Major contributors to the development of pancreatic cancer were of particular emphasis, namely TP53, KRAS, CDKN2a, and SMAD4 [2, 3]. We checked the sequences generated from Panc268, Panc269, Panc320, and respective organoids for mutations in these genes. Table 1 shows the pathogenic mutations identified in accordance with ClinVar [40]. Clinical mutational profiling of PTOL tumors revealed the same frequency of oncogene (e.g. KRAS) and tumor suppressor gene (e.g. TP53) mutations as the general pancreatic cancer population (Table 1). For example, Panc269 expresses common KRAS and TP53 gene mutations whereas Panc272 has no detectable gene mutations. This did not correlate with niche factor dependency determined below. As emphasized by this study, the genotype and biologic phenotypes are difficult to associate to allow for accurate correlation [6]. While we have adapted the utilization of organoids to help bridge this gap between genotype and phenotype [29], this gap remains a challenge when applying in vitro data to a more clinical context. We were able to effectively classify the phenotypes of our PDAC organoids in light of Wnt signaling and response to inhibition and chemotherapy. The question of relation to genotype remained a key question when considering the mechanism behind our data. In addition to the PCR data obtained, heatmapping of Wnt dependent and Wnt independent genes confirmed Wnt independent gene signatures for most Wnt independent organoids, and Wnt dependent gene signatures for most Wnt dependent organoids, again consistent with previous work done by Satos group [1]. Consistent gene signatures that could be used to more efficiently classify Wnt (in)dependency in pancreatic tumors. This classification of gene signatures of PDAC PTOLs could be utilized as a prognostic indicator for consideration of Wnt inhibition therapy in combination with standard chemotherapy for those tumors dependent on Wnt signaling for growth. It could additionally serve as a tool to better predict expected phenotypes for patient derived pancreatic organoids that are developed. Furthermore, we need to validate the differences seen between the organoid and its complimentary PDX. This may include introducing various growth factors to the mouse to compensate for the differences seen in the tumor microenvironment vs the growth media for the organoid. This study was largely limited by the size of our PDAC organoid library and the utility of these organoids dictated by varying growth rates. For more universal acceptance of results, these experiments will need to be conducted on more organoids as they are developed. |